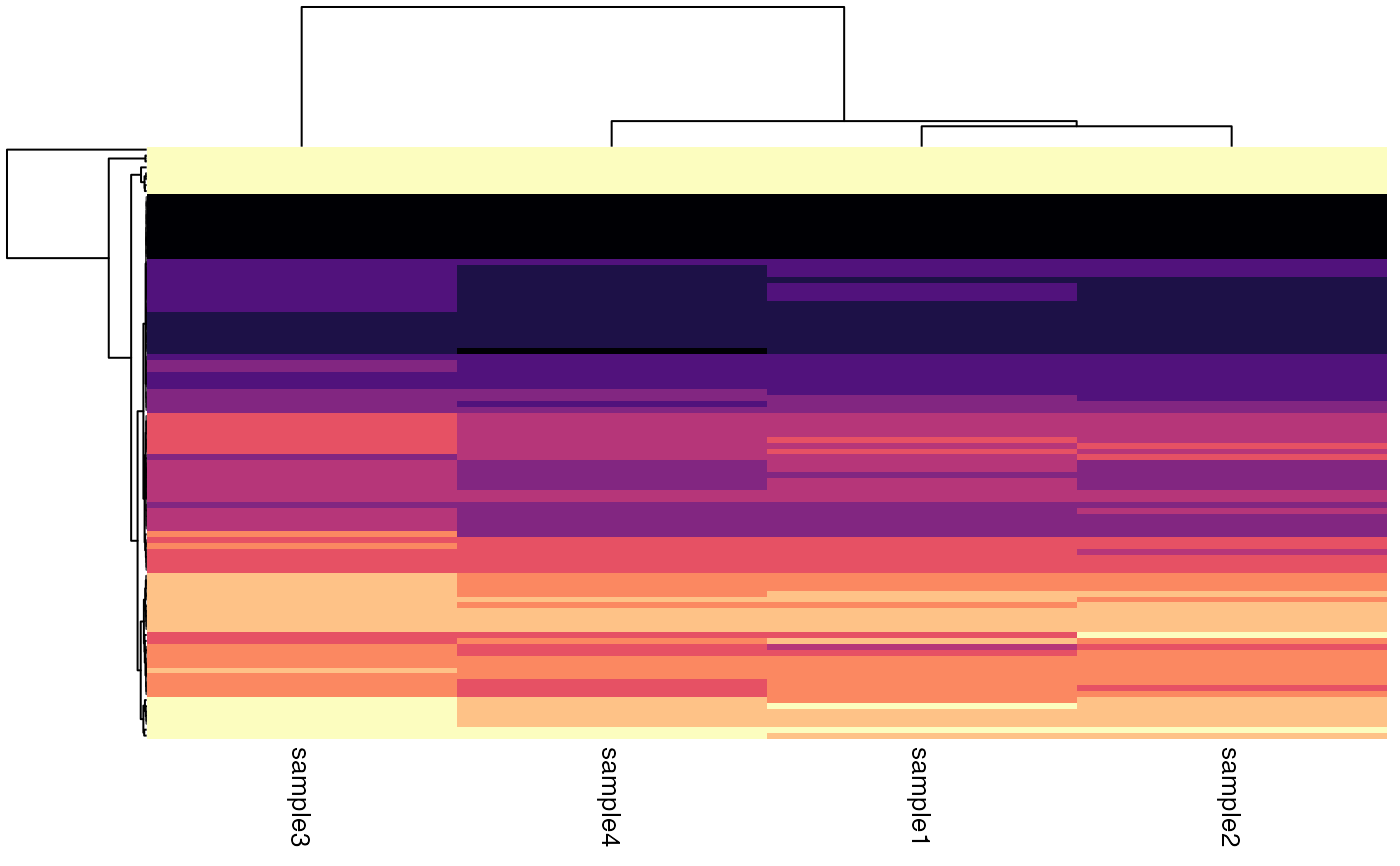Quantile heatmap
Usage
plotQuantileHeatmap(object, ...)
# S4 method for class 'SingleCellExperiment'
plotQuantileHeatmap(object, ...)
# S4 method for class 'SummarizedExperiment'
plotQuantileHeatmap(
object,
assay = 1L,
interestingGroups = NULL,
n = 10L,
clusterRows = TRUE,
clusterCols = TRUE,
showRownames = isTRUE(nrow(object) <= 30L),
showColnames = TRUE,
treeheightRow = 50L,
treeheightCol = 50L,
color = getOption(x = "acid.heatmap.quantile.color", default = viridis::magma),
legendColor = getOption(x = "acid.heatmap.legend.color", default =
AcidPlots::synesthesia),
legend = FALSE,
borderColor = NULL,
title = NULL,
convertGenesToSymbols = showRownames,
...
)Arguments
- object
Object.
- ...
Additional arguments.
- assay
vector(1). Assay name or index position.- interestingGroups
character. Groups of interest to use for visualization. Corresponds to factors describing the columns of the object.- n
integer(1). The number of quantile breaks to create.- clusterRows, clusterCols
logical(1). Arrange with hierarchical clustering.- showRownames, showColnames
logical(1). Show row or column names.- treeheightRow, treeheightCol
integer(1). Size of the row and column dendrograms. Use0to disable.- color
function,character, orNULL. Hexadecimal color function or values to use for plot.We generally recommend these hexadecimal functions from the viridis package, in addition to our
synesthesia()palette:Alternatively, colors can be defined manually using hexadecimal values (e.g.
c("#FF0000", "#0000FF")), but this is not generally recommended. Refer to the RColorBrewer package for hexadecimal color palettes that may be suitable. If setNULL, will use the default pheatmap colors.- legendColor
functionorNULL. Hexadecimal color function to use for legend labels. Note that hexadecimal values are not supported. If setNULL, will use the default pheatmap colors.- legend
logical(1). Show the color legend.- borderColor
character(1)orNULL. Border color.- title
character(1). Title.- convertGenesToSymbols
logical(1). Attempt to automatically convert gene identifiers to gene symbols. Only applies when object contains mappings defined inrowRanges.
Examples
data(
RangedSummarizedExperiment,
SingleCellExperiment_splatter,
package = "AcidTest"
)
## SummarizedExperiment ====
object <- RangedSummarizedExperiment
plotQuantileHeatmap(object)
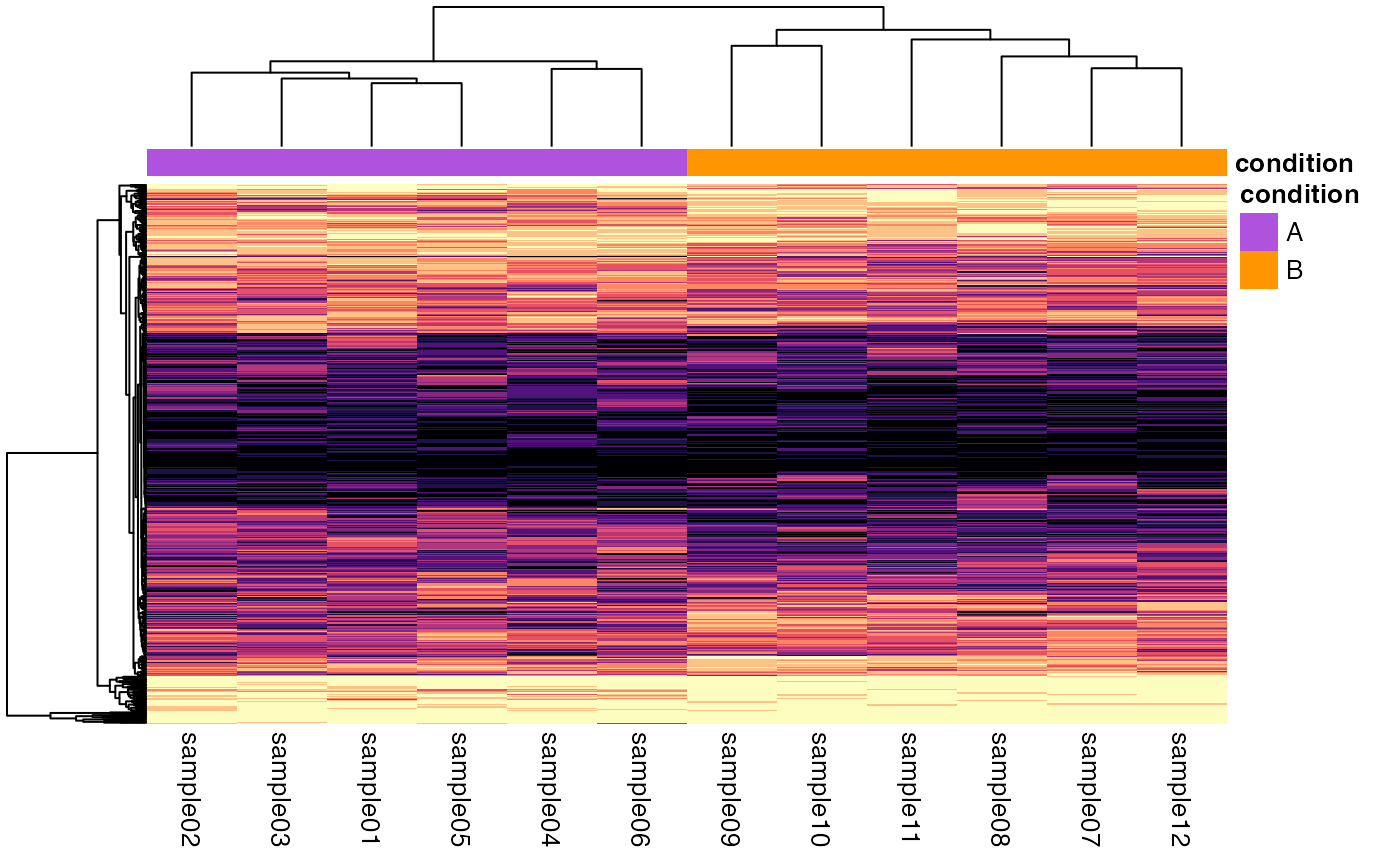 ## SingleCellExperiment ====
object <- SingleCellExperiment_splatter
plotQuantileHeatmap(object)
## SingleCellExperiment ====
object <- SingleCellExperiment_splatter
plotQuantileHeatmap(object)