Plot ribosomal RNA (rRNA) mapping rate
Source:R/AllGenerics.R, R/plotRrnaMappingRate-methods.R
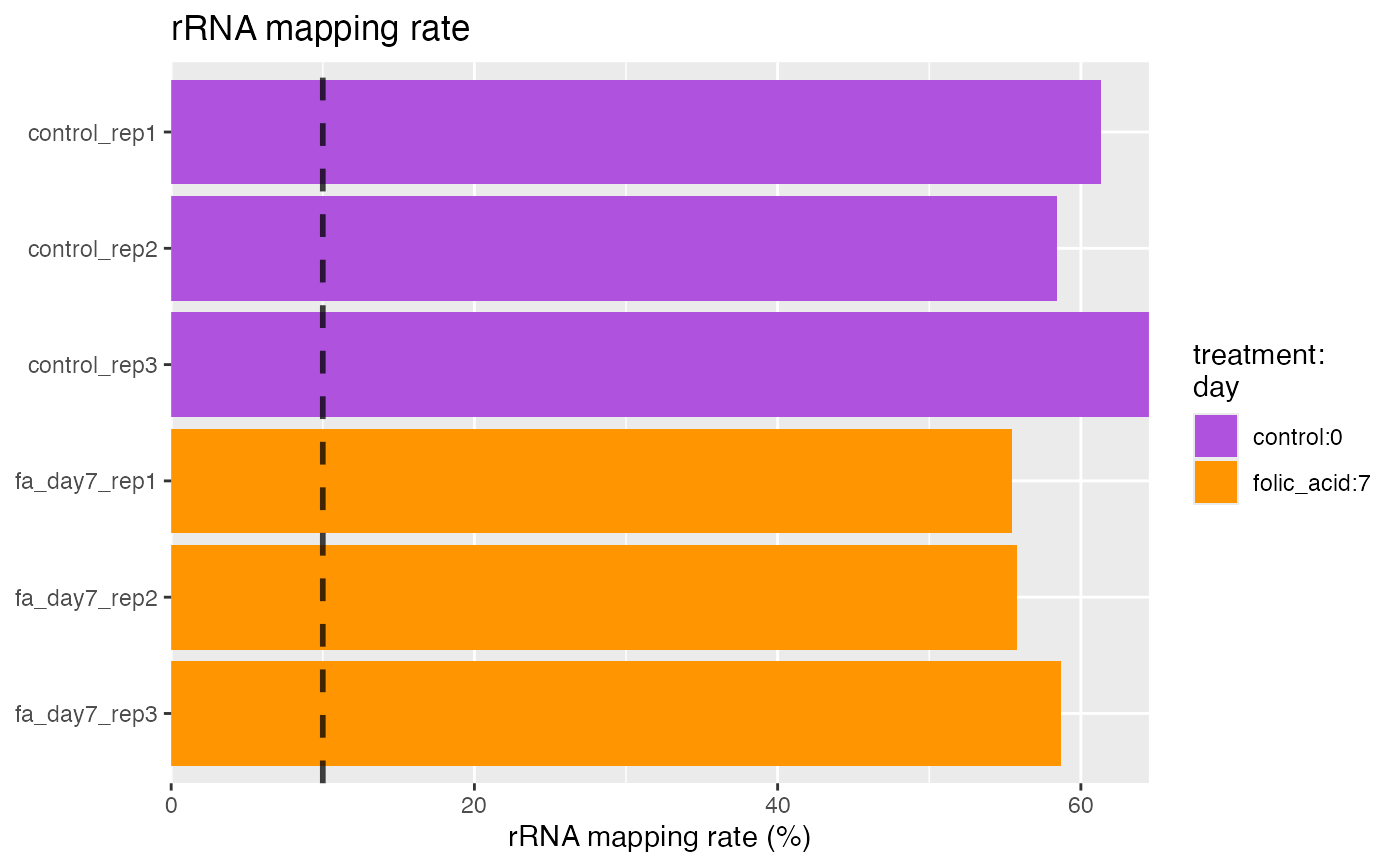
plotRrnaMappingRate.RdClean, high-quality samples should have an rRNA mapping rate below 10%. Higher rates are likely indicative of the polyA enrichment or ribo depletion protocol not having removed all ribosomal RNA (rRNA) transcripts. This will reduce the number of biologically meaningful reads in the experiment and is best avoided.
Arguments
- object
Object.
- interestingGroups
character. Groups of interest to use for visualization. Corresponds to factors describing the columns of the object.- limit
numeric(1). Threshold limit.- labels
list. ggplot2 labels. Seeggplot2::labs()for details.- flip
logical(1). Flip x and y axes. Recommended for plots containing many samples.- ...
Additional arguments.
Examples
data(bcb)
## bcbioRNASeq ====
plotRrnaMappingRate(bcb)