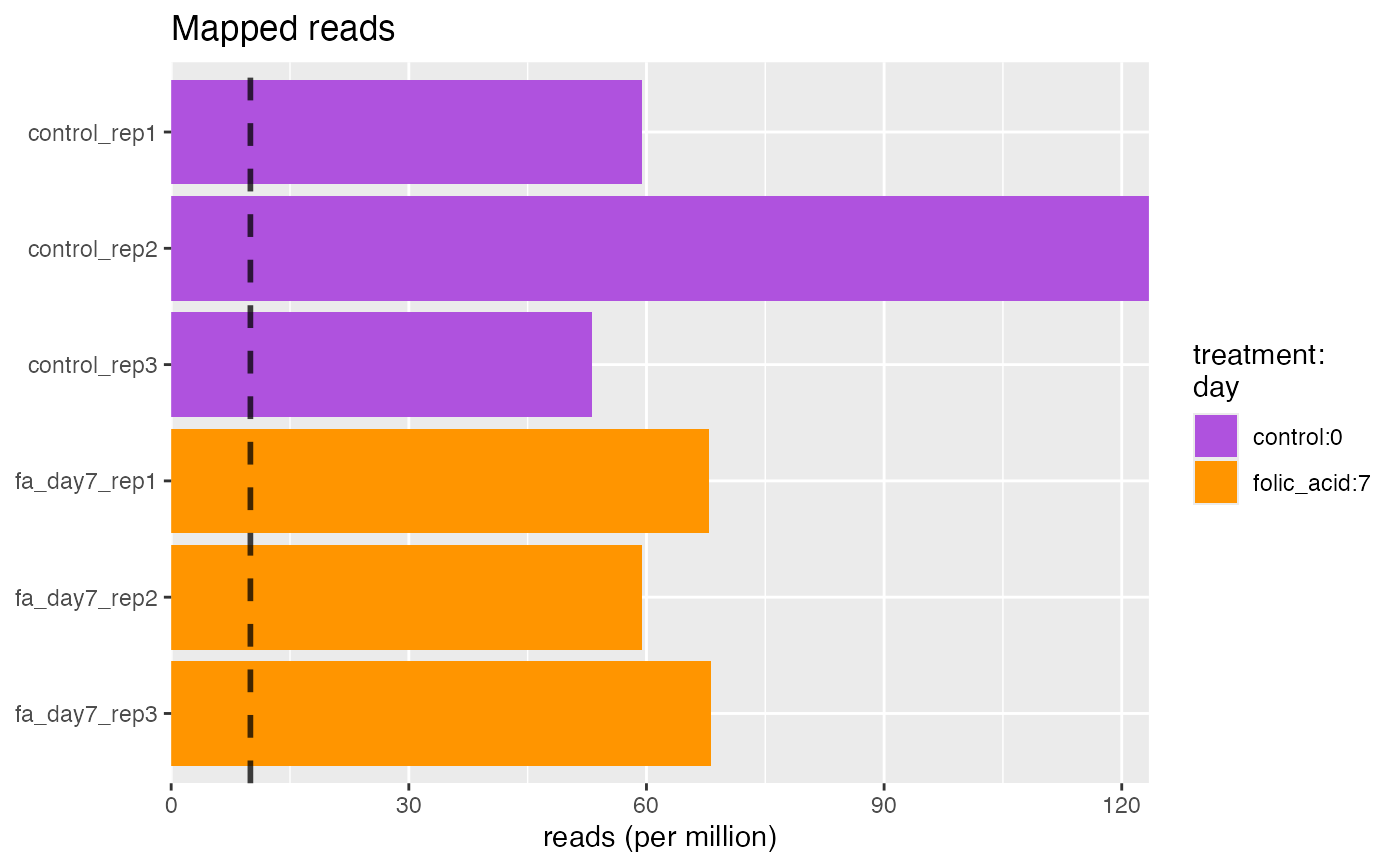
The number of mapped reads should correspond to the number of total reads.
Arguments
- object
Object.
- interestingGroups
character. Groups of interest to use for visualization. Corresponds to factors describing the columns of the object.- limit
numeric(1). Threshold limit.- perMillion
logical(1). Display as counts per million.- labels
list. ggplot2 labels. Seeggplot2::labs()for details.- flip
logical(1). Flip x and y axes. Recommended for plots containing many samples.- ...
Additional arguments.
Examples
data(bcb)
## bcbioRNASeq ====
plotMappedReads(bcb)