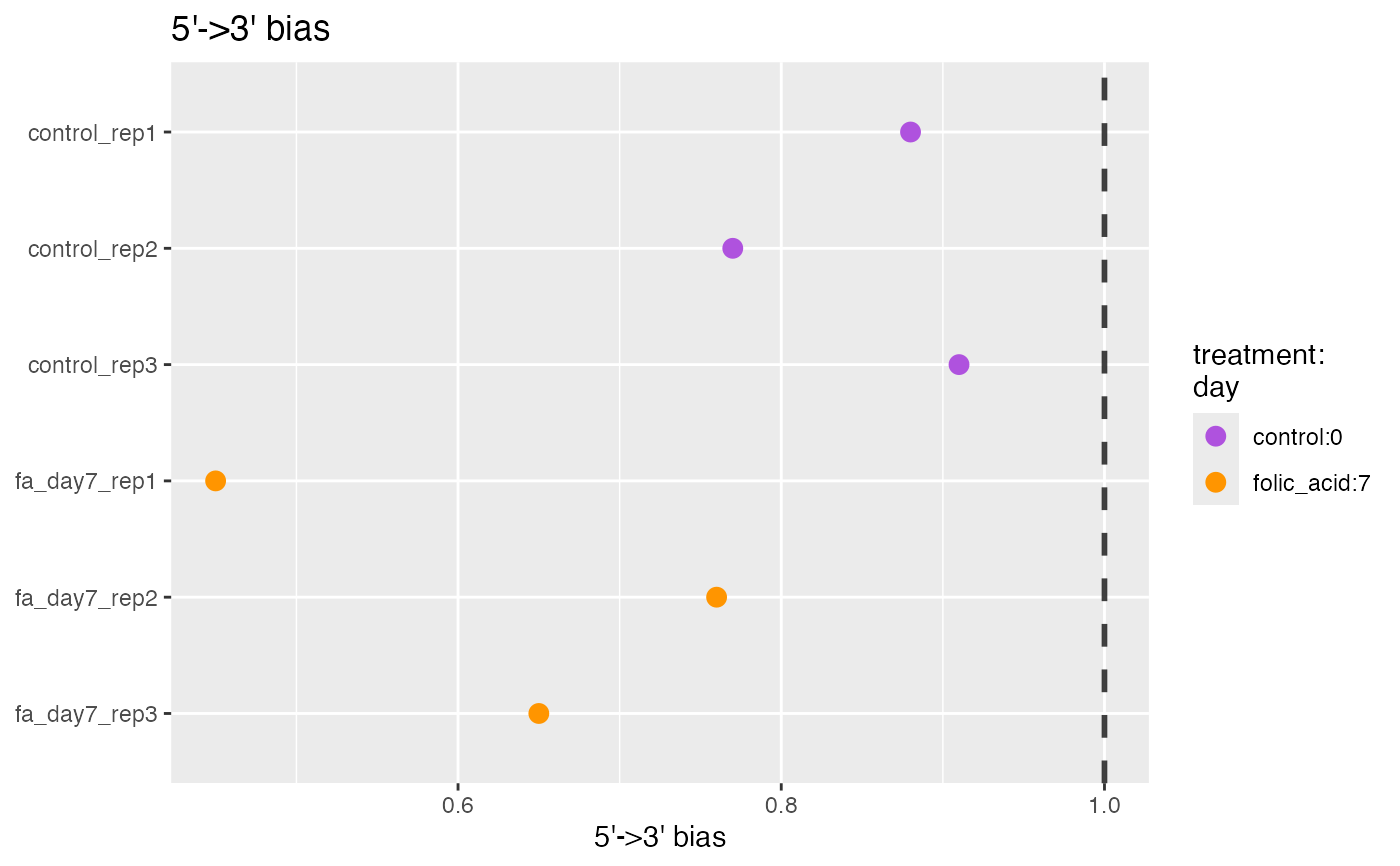
Plot 5' to 3' bias
Source:R/AllGenerics.R, R/plot5Prime3PrimeBias-methods.R
plot5Prime3PrimeBias.RdRNA-seq data can have specific biases at either the 5’ or 3’ end of sequenced fragments.
Arguments
- object
Object.
- interestingGroups
character. Groups of interest to use for visualization. Corresponds to factors describing the columns of the object.- labels
list. ggplot2 labels. Seeggplot2::labs()for details.- flip
logical(1). Flip x and y axes. Recommended for plots containing many samples.- ...
Additional arguments.
Details
It is common to see a small amount of bias, especially if polyA enrichment was performed, or if there is any sample degradation. If a large amount of bias is observed here, be sure to analyze the samples with a Bioanalyzer and check the RIN scores.
5' (3') bias is generally calculated as the median of the following ratio:
For example:
Mean expression for 5' (3') is calculated as mean coverage of first (last) 100 bases.
Mean expression of transcript is the mean coverage of all bases in that transcript.
Median is calculated for the representative set of 1000 transcripts.
Examples
data(bcb)
## bcbioRNASeq ====
plot5Prime3PrimeBias(bcb)