Plot features detected
Source:R/AllGenerics.R, R/plotFeaturesDetected-methods.R
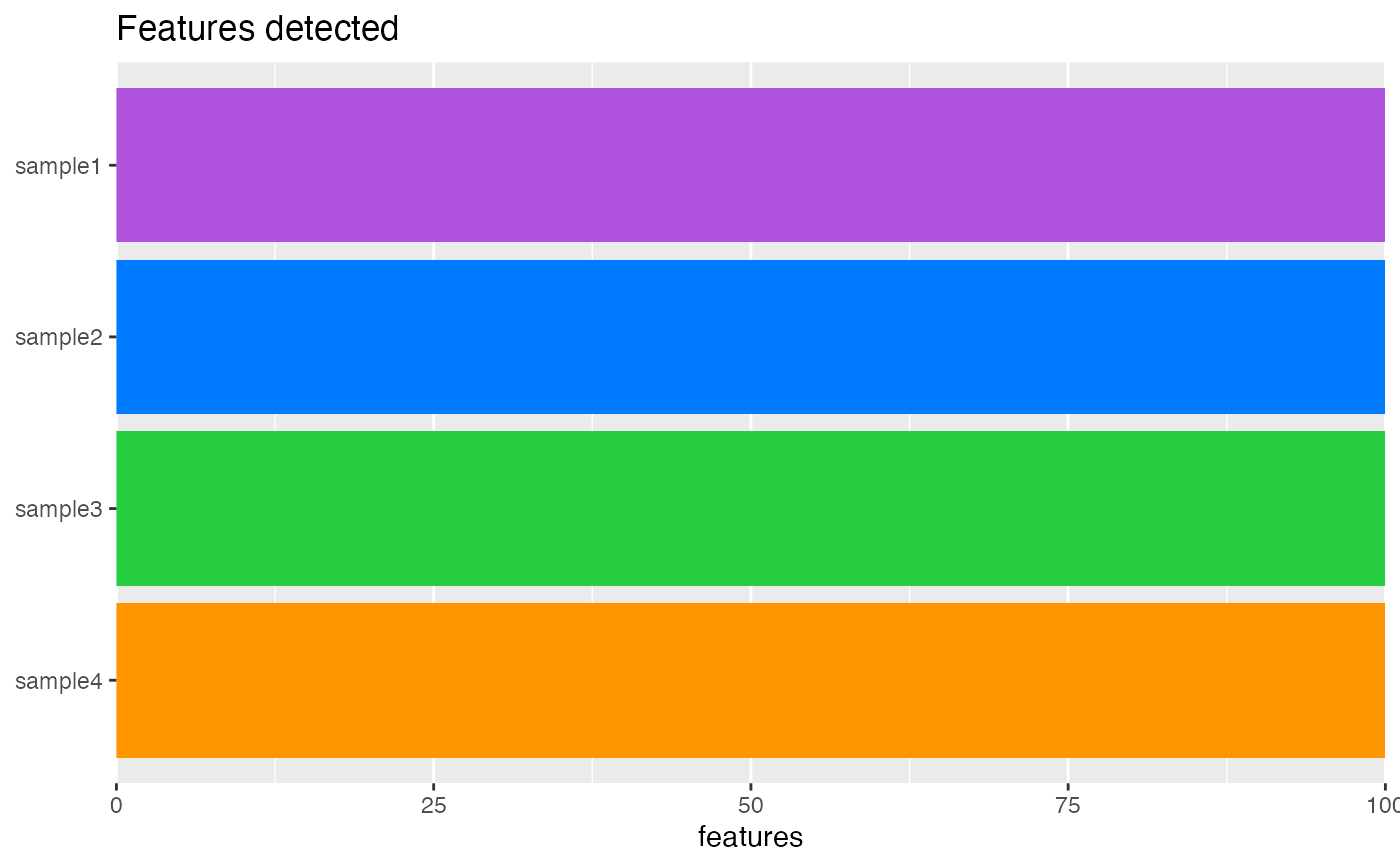
plotFeaturesDetected.RdVisualize the number of features (i.e. genes) detected.
Usage
plotFeaturesDetected
# S4 method for class 'SingleCellExperiment'
plotFeaturesDetected(object, ...)
# S4 method for class 'SummarizedExperiment'
plotFeaturesDetected(
object,
assay = 1L,
interestingGroups = NULL,
limit = 0L,
minCounts = 1L,
labels = list(title = "Features detected", subtitle = NULL, x = NULL, y = "features"),
flip = getOption(x = "acid.flip", default = TRUE)
)Arguments
- object
Object.
- ...
Additional arguments.
- assay
vector(1). Assay name or index position.- interestingGroups
character. Groups of interest to use for visualization. Corresponds to factors describing the columns of the object.- limit
numeric(1). Threshold limit.- minCounts
integer(1). Minimum number of counts per feature (i.e. gene).- labels
list. ggplot2 labels. Seeggplot2::labs()for details.- flip
logical(1). Flip x and y axes. Recommended for plots containing many samples.
Functions
plotFeaturesDetected(SingleCellExperiment): AppliesaggregateCellsToSamples()calculation to summarize at sample level prior to plotting.
Passes...toSummarizedExperimentmethod.
Examples
data(
RangedSummarizedExperiment,
SingleCellExperiment_splatter,
package = "AcidTest"
)
## SummarizedExperiment ====
object <- RangedSummarizedExperiment
plotFeaturesDetected(object)
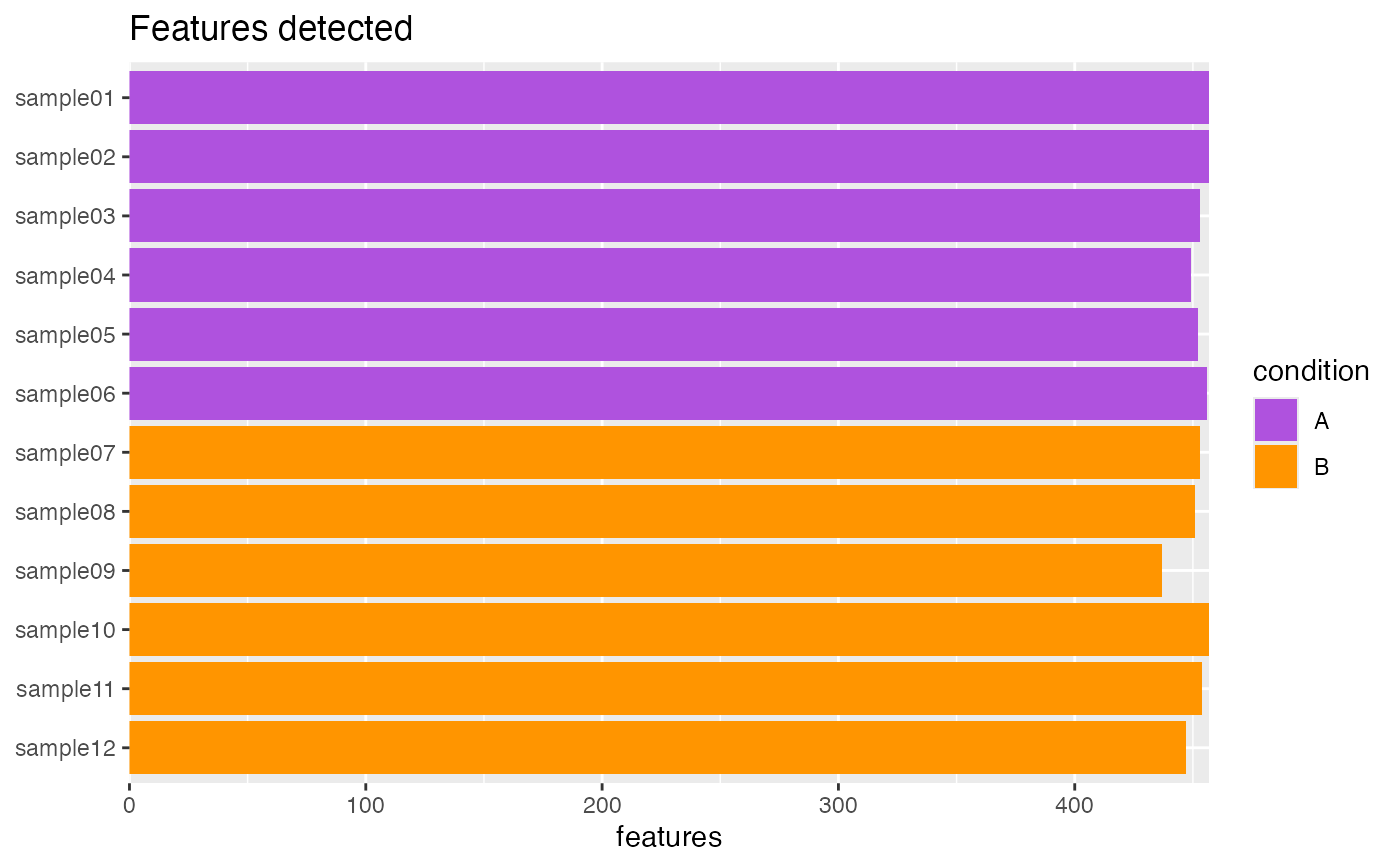 ## SingleCellExperiment ====
object <- SingleCellExperiment_splatter
plotFeaturesDetected(object)
## SingleCellExperiment ====
object <- SingleCellExperiment_splatter
plotFeaturesDetected(object)