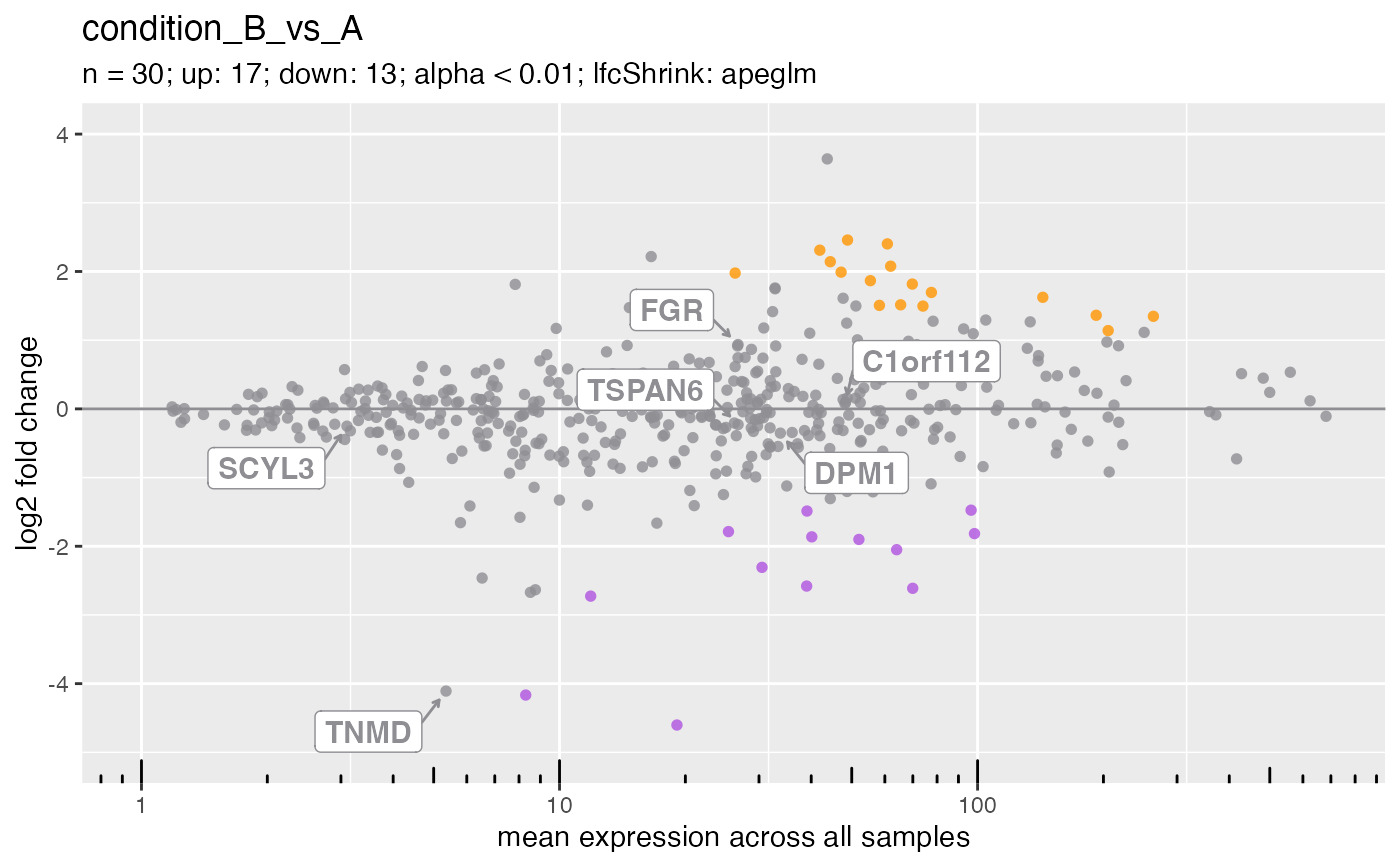
The plot visualizes the differences between measurements taken in two samples, by transforming the data onto M (log ratio) and A (mean average) scales.
Usage
plotMa(object, ...)
# S4 method for class 'DESeqAnalysis'
plotMa(
object,
i,
alphaThreshold = NULL,
baseMeanThreshold = NULL,
lfcThreshold = NULL,
genes = NULL,
ntop = 0L,
...
)
# S4 method for class 'DESeqResults'
plotMa(
object,
direction = c("both", "up", "down"),
alphaThreshold = NULL,
baseMeanThreshold = NULL,
lfcThreshold = NULL,
genes = NULL,
ntop = 0L,
pointColor = c(downregulated = AcidPlots::lightPalette[["purple"]], upregulated =
AcidPlots::lightPalette[["orange"]], nonsignificant =
AcidPlots::lightPalette[["gray"]]),
pointSize = 2L,
pointAlpha = 0.8,
limits = list(x = NULL, y = NULL),
labels = list(title = TRUE, subtitle = NULL)
)Arguments
- object
Object.
- i
Indices specifying elements to extract or replace. Indices are
numericorcharactervectors, empty (missing), orNULL.For more information:
- alphaThreshold
numeric(1)orNULL. Adjusted P value ("alpha") cutoff. If leftNULL, will use the cutoff defined in the object.- baseMeanThreshold
numeric(1)orNULL. Base mean (i.e. average expression across all samples) threshold. If leftNULL, will use the cutoff defined in the object. Applies in general to DESeq2 RNA-seq differential expression output.- lfcThreshold
numeric(1)orNULL. Log (base 2) fold change ratio cutoff threshold. If leftNULL, will use the cutoff defined in the object.- genes
character. Gene identifiers.- ntop
integer(1). Number of top features to label.- ...
Additional arguments.
- direction
character(1). Include"both","up", or"down"directions.- pointColor
character(1). Default point color for the plot.- pointSize
numeric(1). Point size for dots in the plot. In the range of 1-3 is generally recommended.- pointAlpha
numeric(1)(0-1). Alpha transparency level.- limits
list(2). Named list containing"x"and"y"that define the lower and upper limits for each axis. Set automatically by default when leftNULL.- labels
list. ggplot2 labels. Seeggplot2::labs()for details.
Details
An MA plot is an application of a Bland–Altman plot for visual representation of genomic data. The plot visualizes the differences between measurements taken in two samples, by transforming the data onto M (log ratio) and A (mean average) scales, then plotting these values.
Functions
plotMa(DESeqAnalysis): Passes toDESeqResultsmethod, withgene2symbolargument automatically defined.
Examples
data(deseq)
## Get genes from DESeqDataSet.
dds <- as(deseq, "DESeqDataSet")
genes <- head(rownames(dds))
print(genes)
#> [1] "gene1" "gene2" "gene3" "gene4" "gene5" "gene6"
## DESeqAnalysis ====
plotMa(deseq, i = 1L)
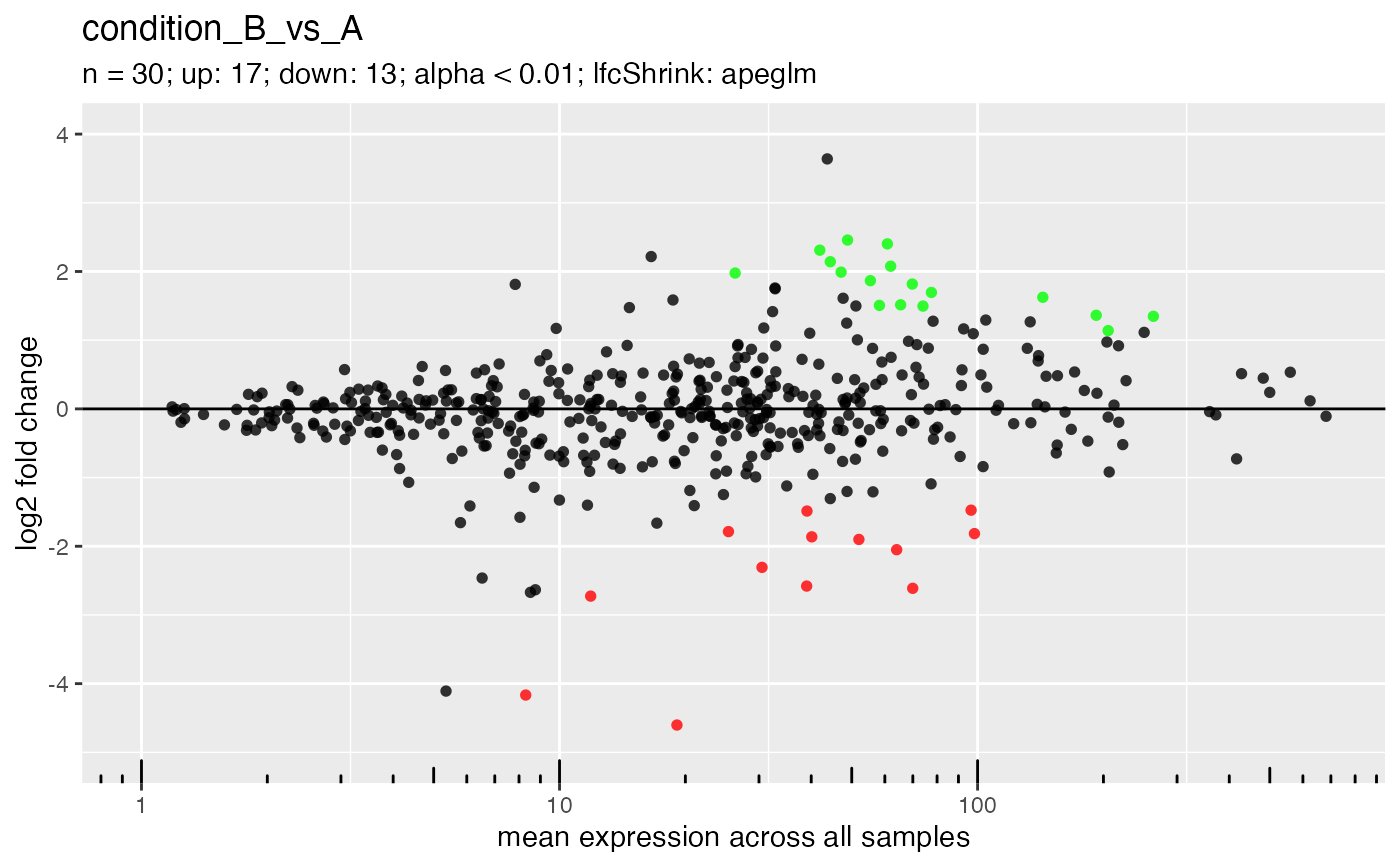
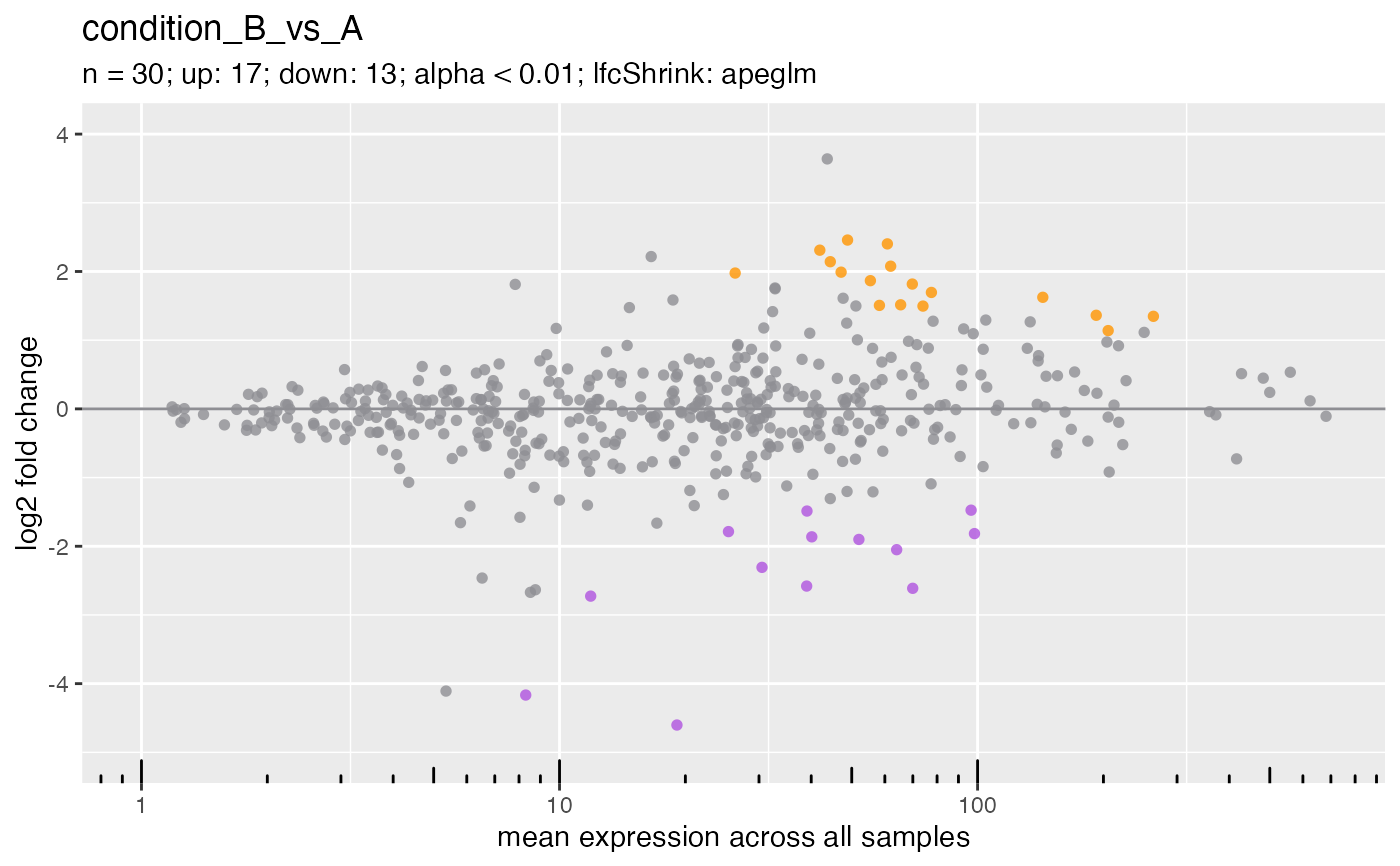 ## Customize the colors.
plotMa(
object = deseq,
i = 1L,
pointColor = c(
downregulated = "red",
nonsignificant = "black",
upregulated = "green"
)
)
## Customize the colors.
plotMa(
object = deseq,
i = 1L,
pointColor = c(
downregulated = "red",
nonsignificant = "black",
upregulated = "green"
)
)
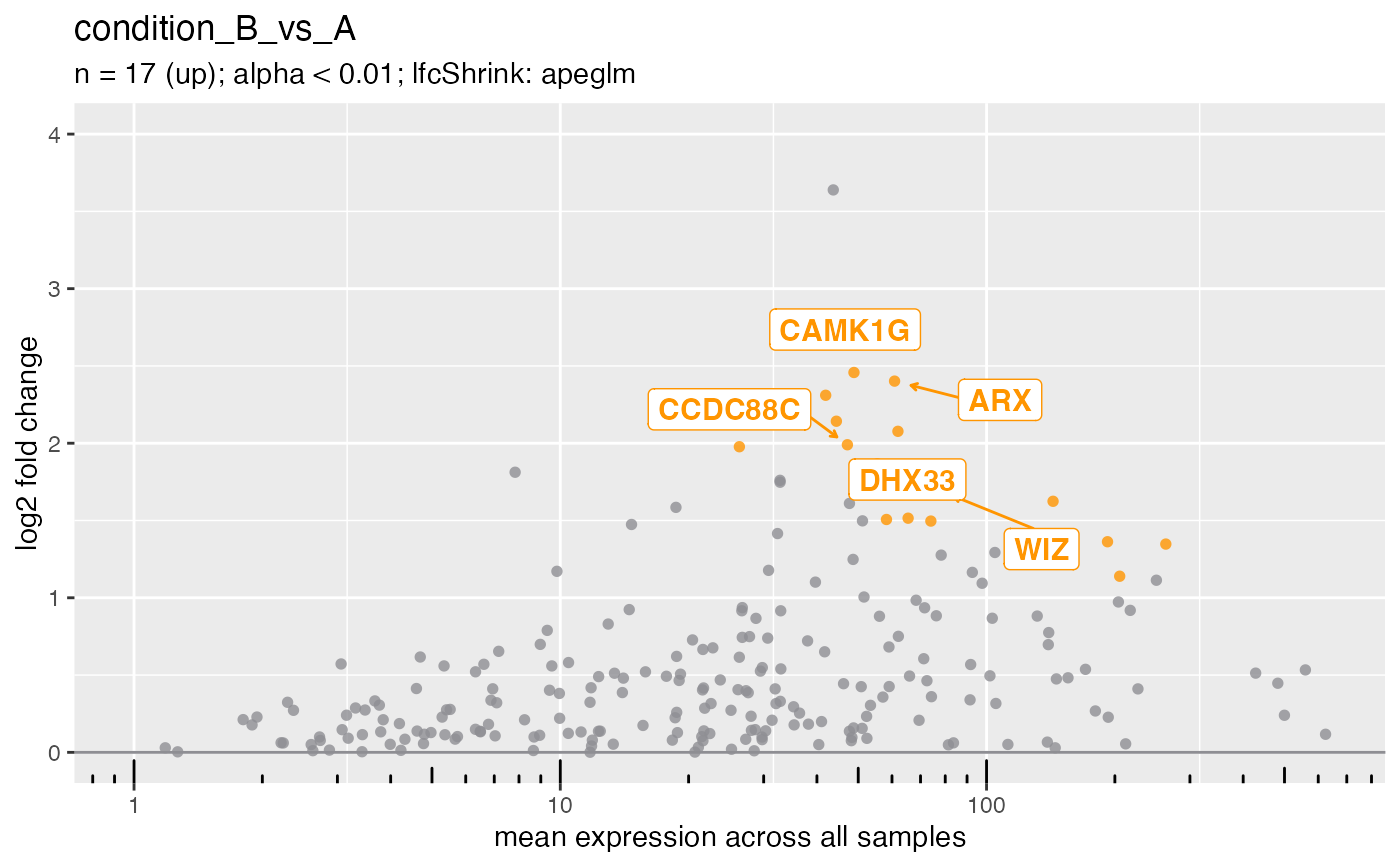
 ## Directional support (up or down).
plotMa(deseq, i = 1L, direction = "up", ntop = 5L)
#> ℹ Labeling 5 genes in plot.
## Directional support (up or down).
plotMa(deseq, i = 1L, direction = "up", ntop = 5L)
#> ℹ Labeling 5 genes in plot.
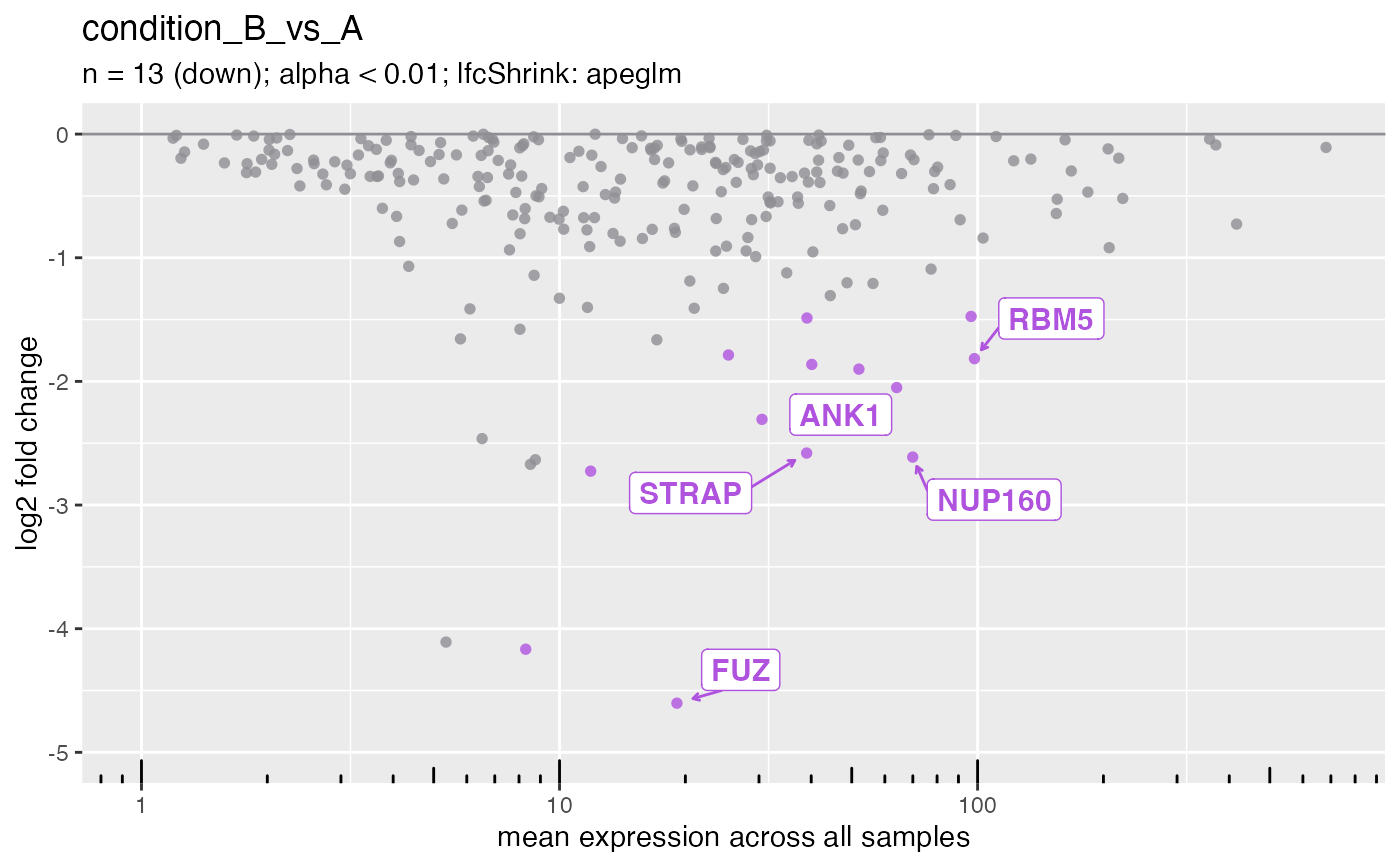
 plotMa(deseq, i = 1L, direction = "down", ntop = 5L)
#> ℹ Labeling 5 genes in plot.
plotMa(deseq, i = 1L, direction = "down", ntop = 5L)
#> ℹ Labeling 5 genes in plot.
 ## Label genes manually.
## Note that either gene IDs or names (symbols) are supported.
plotMa(deseq, i = 1L, genes = genes)
#> ℹ Labeling 6 genes in plot.
## Label genes manually.
## Note that either gene IDs or names (symbols) are supported.
plotMa(deseq, i = 1L, genes = genes)
#> ℹ Labeling 6 genes in plot.